A Genome-Wide Association Study of Bipolar Disorder Using a Subphenotype: Sleeplessness Symptom of Bipolar Mania
Article information

Abstract
Objective
Dividing bipolar disorder (BD) into clinical subphenotypes is a possible approach to determine the genetic basis of BD. The goal of this study was to find genes associated with BD by performing a genome-wide association study (GWAS) based on the subphenotypes, i.e., sleeplessness symptom of bipolar mania (SBM) vs. non-sleeplessness symptom of bipolar mania (NSBM).
Methods
A total of 2,191 cases, 1,434 controls, and 703,012 single nucleotide polymorphisms (SNPs) in the merged samples from the Translational Genomics Institute and the Genetic Association Information Network were investigated. In individuals with bipolar I disorder, a GWAS of SBM vs. NSBM was performed.
Results
We identified 44 trait-associated SNPs with p<10-4 in a case-only analysis. The most statistically significant peak p-value was observed for rs10492908 (odds ratio=0.1832; p=9.27×10-7; Permuted p=1.00×10-6), which is located within the region of the genome encoding WW domain-containing oxidoreductase (WWOX) on chromosome 16q23. A total of eight genomic regions of interest (ROIs) on chromosomes 3, 5, 8, 9, 12, and 17 were also identified, and ROR2 and ANKFN1 in two ROIs may be related with SBM.
Conclusion
Our results suggest several candidate genes and pathways that are related to SBM. In particular, WWOX, ROR2, and ANKFN1 are candidate genes related to SBM. Additional replication studies are needed to confirm these results.
INTRODUCTION
Bipolar disorder (BD) is a common mental disorder characterized by episodic mood symptoms, including episodes of mania or hypomania and depression, and its mood episodes are severe and lifelong [1,2]. The lifetime prevalence of BD is approximately 3.9–6.4% in the general population [3-5], and the lifetime prevalence of bipolar I disorder (BID) is between 1% and 2% [6,7]. Relapse and recurrence are very common and the lifetime risk of suicide is as high as 20% in BD patients, resulting in serious economic costs [8].
The representative defining character of BD is the development of alternating episodes of mania, hypomania, or depression [6]. All BD episode types typically show prominent sleep disturbance. Usually, the necessity for sleep is reduced during mania or hypomania episodes, and patients typically suffer from insomnia or hypersomnia during major depressive episodes [9]. Several studies have suggested that sleep deprivation results in significantly increased negative mood [10,11]. Sleep disturbances may contribute to mood-related features across psychiatric disorders [12]. A bidirectional relationship between sleep dysfunction and symptoms of psychiatric disorders has been hypothesized, such that a vicious cycle of disturbance in mood and symptoms interrupt nighttime sleep, and this sleep deprivation contributes to mood regulation difficulties and symptom worsening [12,13]. Several findings have suggested that circadian rhythm dysfunction is associated with the pathophysiology of BD [14-17]. Commonly, BD patients have circadian rhythm-related clinical manifestations, such as a cyclical/seasonal pattern of relapse, activity changes, periodicity of manic-depressive episodes, diurnal variation in mood, alteration of the secretion of hormones and other endogenous substances, as well as sleep disturbances, such as hypersomnia or insomnia during depression and sleeplessness during mania [18]. Additionally, the interpersonal, environmental, and pharmacological events of BD may cause sleep deprivation [13,19]. Sleep disturbances can be caused by circadian dysfunctions in BD, and inversely, sleep disturbances may have an influence on emotional regulation [20]. Our group previously suggested circadian rhythm hypotheses to explain mixed features, antidepressant treatment resistance, and manic switching in BD, and we emphasized that circadian dysregulation may cause recurrent unipolar depression and BD [21-25]. Among the disturbance of sleep-wake rhythm, sleeplessness—reduced need for sleep—is a distinctive symptom in the bipolar mania. In the diagnostic criteria of BD, ‘decreased need for sleep’ is one of the main symptoms of the mania or hypomania [26]. Thus, the sleeplessness is worthy of consideration as a major subphenotype of bipolar mania.
Even though BD is a highly heritable psychiatric disorder, the investigation of specific genetic variations has suggested limited findings [27-30]. Since, it has been suggested that dividing BD into subgroups according to clinical phenotypes is a possible approach for further meaningful genetic studies in BD. Lett et al. [31] reported that ZNF804A and CACNA1C are associated with a psychotic subphenotype of BD. Our group previously performed a genome-wide association (GWA) analysis of BD using subphenotypes, such as seasonal pattern mania [32] and hypersomnia symptom of bipolar depression [33].
In the present study, we performed a GWA analysis of the sleeplessness symptom of bipolar mania (SBM) vs. non-sleeplessness symptom of bipolar mania (NSBM) in BD subjects and controls of European ancestry, genotyped as part of the Genetic Association Information Network (GAIN) by the Bipolar Genome Study (BiGS).
METHODS
Subject ascertainment
A total of 1,556 unrelated BID subjects of European ancestry were selected from those collected by the National Institute of Mental Health Genetics Initiative for Bipolar Disorder in 5 waves at 11 sites across the United States. Recruitment for waves 1–2 consisted of extended multiplex families with a BID proband, waves 3–4 included families with a BID proband and at least one other sibling with BID or schizoaffective disorder, bipolar type, and wave 5 consisted of unrelated BID cases. All subjects provided written informed consent according to the local Institutional Review Board protocols and were interviewed using the Diagnostic Interview for Genetic Studies (DIGS) [34]. Information sources for the study included both family informants and medical records. These information sources were reviewed in addition to the interview by a panel of experienced clinicians to reach a final best-estimate diagnosis. The full version of the DIGS interview contains over 2,000 questions with detailed information regarding manic and depressive episodes, including sleep patterns, and is available for these subjects. Control subjects were selected from those confirmed based on a National Institute of Mental Health-supported contract mechanism between Dr. Pablo Gejman and Knowledge Networks, Inc [35]. All subjects donated a blood sample and completed a medical questionnaire. The controls were matched with respect to ethnicity and gender to the BD cases, and all control subjects who had a history of BD, psychosis, or recurrent major depression were excluded from our study.
Genotyping and cleaning
Initial samples were genotyped at the Broad Institute as part of GAIN using Affymetrix SNP Array 6.0 [Affymetrix, Santa Clara, CA, USA; 1M single nucleotide polymorphism (SNPs)]. A total of 1,001 BD cases, 1,033 controls, and 724,067 SNPs were available for analysis following an extensive quality control process to eliminate individuals with >10% missing data, SNPs with poor allele clustering, duplicate data, significant deviation from Hardy–Weinberg equilibrium at p<10-6, and minor allele frequencies <0.05. The second sample was genotyped at the Translational Genomic Institute (TGEN) and a comparable quality control process was used, resulting in 1,190 BD cases, 401 controls, and 728,187 SNPs for analysis. A total of 2,191 cases, 1,434 controls, and 703,012 SNPs in the merged samples of TGEN and GAIN were investigated.
Phenotypes
Phenotypes were based on the information from the Bipolar Disorder Phenome Database, which is a resource for genetic studies that compiles data across the DIGS 2, 3, and 4 to obtain a common set of variables for samples from each subject [32,36]. As a part of the DIGS interview, BID subjects were queried about sleep during the most severe manic episode. Subjects were asked the question, “Did you need less sleep than usual?’’ BID subjects who answered ‘Yes’ were assigned to the SBM group and subjects who answered ‘No’ were assigned to the NSBM group. Those who answered ‘Unknown’ to the question were categorized as missing because their symptoms may be not related to sleep patterns. After categorization, there were 302 BID subjects in the SBM group and 84 subjects in the NSBM group.
Association analyses
To investigate the genetic factors contributing to SBM, a caseonly analysis of SBM vs. NSBM was performed. This association analysis was performed using logistic regression with the PLINK software package [37] and covariate adjustment for age and sex. Because a sampling bias owing to the selection of a small subset of individuals may result in spurious findings, adaptive permutations were performed to assess the significance of the results using PLINK. After SNP imputation, the same logistic regression analysis was repeated using the imputed sample for the functional enrichment analysis. A genome-wide significance threshold of p< 5×10-8 was used after correction for multiple testing issues [38].
Linkage disequilibrium analysis
Common linkage disequilibrium (LD) blocks were identified using the Haploview software based on D’ and r2, with default parameters and HapMap CEU+TSI(R2) [39]. Furthermore, proxy SNPs based on regional recombination rates were identified using SNAP and 1000 Genomes Pilot 1 data.
SNP imputation
The genotypes for missing markers in a dataset can be extrapolated on the basis of the correlation between genotypes in a reference dataset and an LD analysis. Similar levels of association should be observed for genotyped markers and imputed markers. The IMPUTE2 tool [40] and the CEU panel of HapMap 3 + 1000 Genomes Pilot haplotypes as a reference were used for imputation.
Functional enrichment analysis
A functional enrichment analysis was performed on the gene sets harboring the identified SNPs (p<0.005) using DAVID software [41]. The gene sets were derived from the results of a logistic regression analysis performed within the imputed sample. An annotation process was performed using the RefSeq database (https://www.ncbi.nlm.nih.gov/refseq/). R software version 3.0 (R Foundation for Statistical Computing, Vienna, Austria) was used for annotation, and each SNP was matched to a gene located within 100,000 base pairs. Significant functions were suggested based on enrichment scores.
RESULTS
We performed a GWA analysis to identify SNPs associated with SBM. Figure 1 shows a Manhattan plot representing the chromosomal view of the log p-values for the SNPs. We identified 44 traitassociated SNPs with p<10-4 in the case-only analysis, and the lowest p-value was observed for rs10492908 [odds ratio (OR)=0.1832; p=9.27× 10-7; Permuted P=1.00×10-6]. This SNP was located within the region of the genome encoding WW domain-containing oxidoreductase (WWOX) on chromosome 16q23.
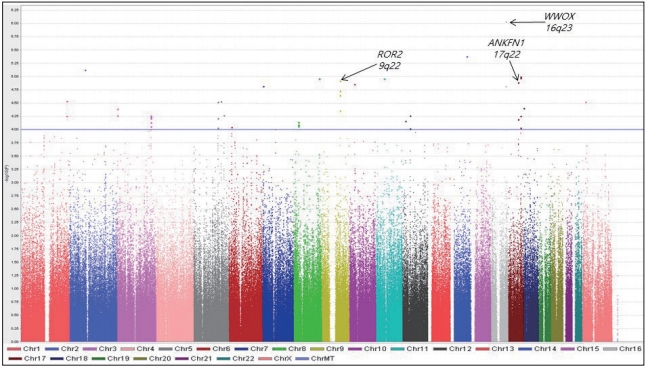
Manhattan plot of genome-wide association results for sleeplessness symptom of bipolar mania susceptibility loci. The chromosomal position is shown along the x-axis, the -log (p-value) for each single nucleotide polymorphism is shown along the y-axis. Horizontal line indicates the p<10-4 threshold.
We identified a total of eight genomic regions of interest (ROIs) on chromosomes 3, 5, 8, 9, 12, and 17 that contained at least two SNPs with p<10-4 and adequate support for an association (i.e., p< 10-3) for additional SNPs within 100 kb. The 5 ROIs, except ROI3 and ROI4, were located in 9 genes. Among these genes, seven included at least 3 SNPs, CNTN4 in ROI1, NCEH1, ECT2 in ROI2, ROR2 in ROI5, ANKFN1 in ROI7, and LINC01483 in ROI8 (Supplementary Table 1 in the online-only Data Supplement).
LD for the ROIs was calculated from HapMap CEU+TSI(R2) using Haploview software after imputing the data using genotypes from the publicly available CEU HapMap 3 + 1000 Genomes Pilot haplotypes data set. As shown in Figure 2, all eight ROIs were located within or near blocks of high LD. These results suggest that these regions have functional significance related to SBM. For the first ROI, rs7646671, which showed a peak p-value of 2.7×10-5, was located within the CNTN4 gene on 3p26.3, and six additional SNPs were observed with p-values of <10-3 within the same region (Figure 2A). In the second ROI, rs11706689, which showed a peak p-value of 4.26×10-5, was located within the ECT2 gene on 3q26, and four additional SNPs were detected in this ROI (p<10-3) (Figure 2B). In ROI6, rs10785430 had the lowest p-value of 4×10-5, and was located within the ADAMTS20 gene on 12q12 and six other SNPs (p<10-3) were detected in the ROI (Figure 2F). Although only a few SNPs within the other 5 ROIs were located in genes, these regions showed significance of p<10-3 in the association study. These results suggest that functional alterations of genes in ROIs may contribute to the occurrence of SBM.
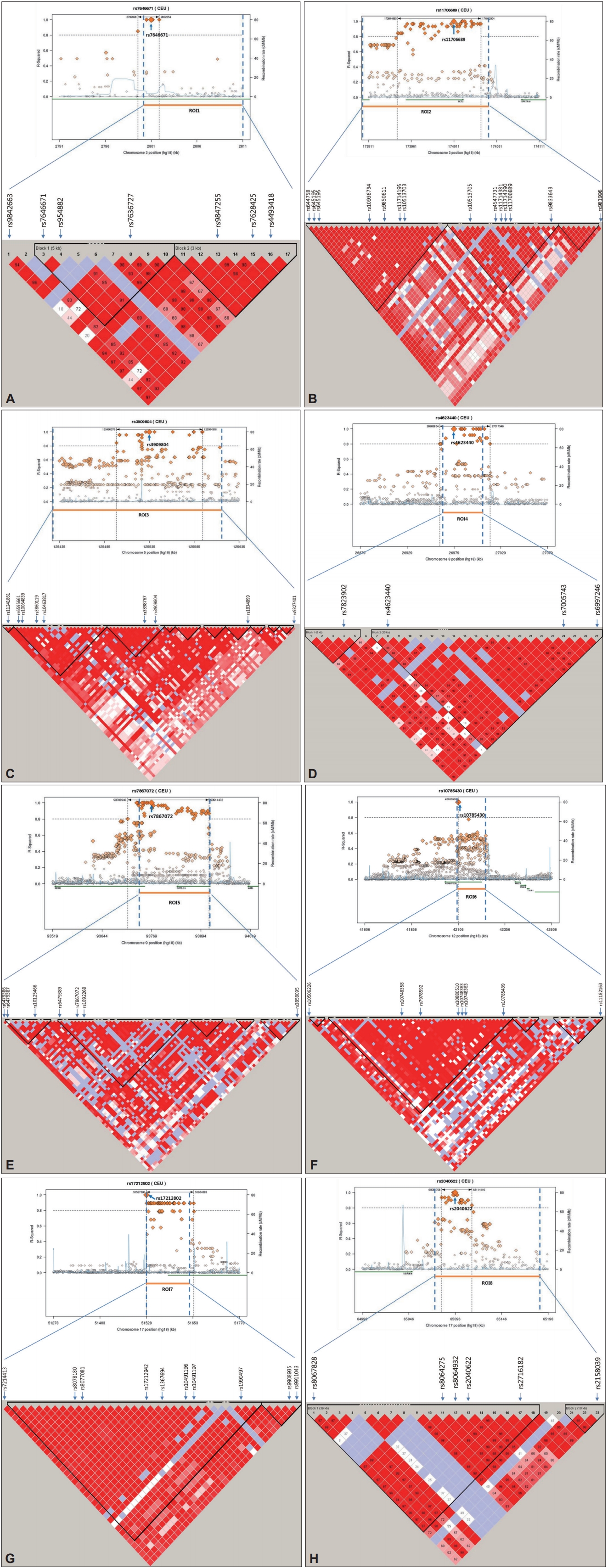
Haplotype association plots for the eight ROIs in the case-control analysis of GWA. Proxy SNPs with regional recombination rates in the eight ROIs are shown as determined by SNAP with HapMap 1000 Genomes Pilot 1 data (A-H, top). The regional LDs around the eight ROIs are shown by Haploview using HapMap CEU+TSI(R2) data (A-H, bottom). The D’ scores for each paired SNP are indicated by the color scheme. The recombination rate (pale blue line), the lowest p-value SNP (the biggest diamond mark), and ROI span (dotted vertical line) are indicated. ROI: region of interest, GWA: genome-wide association, SNP: single nucleotide polymorphism, LD: linkage disequilibrium.
We performed a functional enrichment analysis for associated SNPs after imputing the data using genotypes from the publicly available CEU HapMap 3 + 1000 Genomes Pilot haplotypes data set. We identified gene ontology functions for 3,078 genes containing SNPs demonstrating associations with p<0.005. As shown in Table 1, significant functional enrichments were observed for cell development, cell adhesion, signal transduction, cell recognition, synaptic transmission, and cell motility-related pathways in the SBM vs. NSBM analysis. These findings suggest that the expression of SBM in BD may be related to altered functions in the pathways of the enriched genes identified in this study.
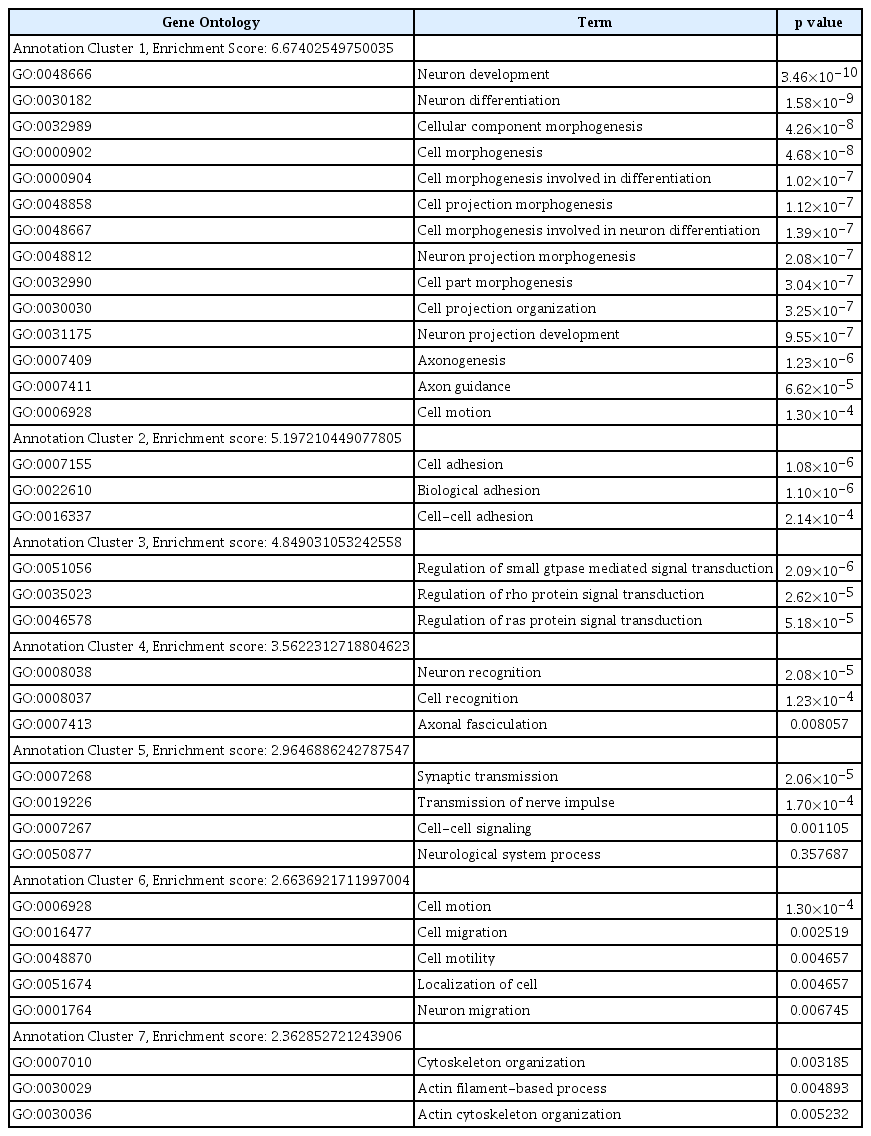
The enrichment findings of the functional enrichment analysis within imputed data of genome-wide association study
DISCUSSION
Various investigations have searched for genetic components contributing to BD susceptibility. Although several GWA studies (GWASs) have suggested possible BD susceptibility genes, there has been little consistency among the results of GWA studies. The Wellcome Trust Case-Control Consortium found an association of rs420259 at chromosome 16p12 (p=6.3×10-8) with BD [42]. Another group found that diacylglycerol kinase eta (DGKH) is associated with BD in a combined GWAS sample (p=1.5×10-8) [43]. MYO5B (p=1.66×10-7) and TSPAN8 (p=6.11×10-7) were suggested to be associated with BD [27]. The Psychiatric GWAS Consortium Bipolar Disorder Working Group confirmed genome-wide significant evidence for an association between BD and CACNA1C (p=1.52× 10-8), which encodes the α subunit of the L-type voltage-gated calcium channel, and ODZ4 (p=4.40×10-8), which encodes a member of a family of cell surface proteins, the teneurins, in a combined GWAS sample [44].
To better understand the genetic basis underlying BD, we performed a GWA study of the SBM subphenotype, which may represent a genetically distinct and homogenous feature. The most statistically significant peak representing a GWA was observed for rs10492908 (OR=0.1832; p=9.27×10-7; Permuted P=1.00×10-6), which is located within WWOX on chromosome 16q23. WWOX has been implicated in human tumorigenesis, especially as a putative tumor suppressor [45]. WWOX has one COOH-terminal short-chain alcohol dehydrogenase/reductase and two N-terminal WW domains (containing conserved tryptophan residues). Since these domains mediate protein–protein interactions, WWOX binds to various proteins. Although WWOX is well-known with regard to cancer, it is also related to nervous system development and Alzheimer’s disease (AD) [46]. Chen et al. [47] reported that WWOX was differentially expressed during various stages of brain development in mice, implicating a potential role for WWOX in promoting neuronal differentiation and maturation. Mallaret et al. [48] showed that WWOX is mutated in autosomal recessive cerebellar ataxia with epilepsy and mental retardation. WWOX is also suggested to have a role in the pathogenesis of AD via tau and glycogen synthase kinase 3β (GSK3β). Tau is a microtubule-associated protein that plays a role in in microtubule assembly and stability. GSK3β has an important role in tau hyperphosphorylation at its microtubule-binding domains, resulting in a disruption of microtubule stability. Wang et al. [49] reported that WWOX can bind directly to GSK3β via its short-chain alcohol dehydrogenase/reductase domain and promotes neuronal differentiation via the negative regulation of GSK3β. GSK3β is a key kinase in several important signaling pathways and is involved in both BD and AD [50].
Lithium is a mood stabilizing medication that is prescribed to control BD. Several studies have suggested that lithium regulates circadian rhythms, and this regulation may mediate the therapeutic effects of lithium [51-53]. A study of chronic lithium administration in rats suggested that lithium influences the synchronization of circadian rhythms by affecting melatonin levels along the retinal-hypothalamic-pineal pathway [54]. Lithium directly and reversibly inhibits GSK3β [55,56]. Treatment of cells with lithium inhibits the GSK3β-dependent phosphorylation of the microtubule-associated protein tau [57]. WWOX, the most statistically significant peak observed in the present GWA study, may be related to BD.
For further evaluation, each gene in the ROIs was reviewed with respect to previously detected associations with psychiatric disorders. We found that receptor tyrosine kinase-like orphan receptor 2 (ROR2) in ROI5 and ANKFN1 in ROI7 may be involved in the sleepless SBM. In mammals, the Ror-family receptor tyrosine kinases include two structurally related proteins, ROR1 and ROR2. ROR1-ROR2 complexes modulate synapse formation in hippocampal neurons [58]. ROR1 is reported to be associated with insomnia [59] and BD [60]. In the developing mouse brain, ROR2 is expressed predominantly in the limbic neocortex, hippocampal neuroepithelium, and caudate putamen, although apparent expression of ROR1 has not been observed in the brain [61]. Ankyrin-repeat and fibronectin type III domain-containing 1 (ANKFN1) in ROI7 is a possible candidate gene based on a GWAS of cannabis dependence [62]. Moreover, we searched for enriched pathways for genes in the ROIs. CNTN4 in ROI1 belonged to all pathways in annotation clusters 1, 2, and 4 and cell motion in annotation cluster 6. ROR2 in ROI5 belonged to all pathways in annotation cluster 2.
Owing to the limitations of genetic studies of psychiatric disorders, we used a subphenotype GWA study to identify candidate genes. The heterogeneous phenotype (BD) was divided into clinically homogeneous subphenotypes, which may be more homogenous at the genetic level as well. Most genetic research related to BD based on subphenotypes has focused on age at onset, psychosis, and episode frequency. Accordingly, we considered SBM as a subphenotype of BD. Although WWOX, which was the most highly significant peak in the GWA did not reach the 5×10-8 threshold for statistical significance in a GWA study and was not observed in the genomic ROIs, this SNP may be associated with BD. A limitation of this study is that candidate genes known to be related to BD from previous studies were not replicated in our results. In the future, genetic studies using more detailed subphenotypes in a large sample of BD are expected to verify the findings of the present study and may suggest more meaningful associations.
Supplementary Materials
The online-only Data Supplement is available with this article at https://doi.org/10.33069/cim.2018.0002.
ROIs with enriched association in the sleeplessness symptom of bipolar mania vs. non-sleeplessness symptom of bipolar mania analysis
Acknowledgements
This study was supported by the Korea Health 21 R&D Project funded by the Ministry of Health & Welfare, Republic of Korea (HM14C2606).
This study data was obtained from the Bipolar Genome Study (BiGS). Especially, we thank to Dr. Kelsoe from UCSD for giving study data.
Notes
The authors have no potential conflicts of interest to disclose.
Author Contributions
Conceptualization: Heon-Jeong Lee. Data curation: Heon-Jeong Lee, Chul-Hyun Cho. Formal analysis: Ji-Hye Choi, Hyun Goo Woo. Investigation: Chul-Hyun Cho, Ji-Hye Choi. Methodology: Ji-Hye Choi, Hyun Goo Woo. Software: Ji-Hye Choi, Hyun Goo Woo. Supervision: Heon-Jeong Lee. Validation: Heon-Jeong Lee, Hyun Goo Woo. Visualization: Chul-Hyun Cho, Ji-Hye Choi. Writing—original draft: Chul-Hyun Cho, Ji-Hye Choi. Writing—review & editing: Heon-Jeong Lee, Hyun Goo Woo.